.png)
Subtyping a complex dementia
A high-resolution view of frontotemporal dementia and TDP-43 accumulation
In nineteenth-century biology, few debates ran hotter than the one between “lumpers” and “splitters”. Lumpers preferred to group organisms into broad categories while splitters preferred to carve out ever more precise distinctions. As a consequence, botanists couldn't even agree on how many species the British flora contained. Joseph Dalton Hooker, one of the era's most prominent naturalists and a committed lumper, found the splitters maddening, convinced their endless naming of new species was not helpful. But his friend Charles Darwin counseled a more measured view. "It is good to have hair-splitters & lumpers," he wrote to Hooker in 1857. That tension has remained a constant in science. It has come to the fore whenever there is a discussion about whether observed differences reflect truly distinct entities or variations on a single theme. It is a tension that runs through the study of frontotemporal lobar degeneration with TDP-43 pathology, or FTLD-TDP.
To take a step back, frontotemporal lobar degeneration (FTLD) is a pattern of tissue degeneration seen within the frontal and temporal lobes of the brain. FTLD is the underlying cause of frontotemporal dementia (FTD), a family of progressive neurodegenerative disorders. FTLD cases, when analyzed at autopsy, sort into three groups, according to the protein that has accumulated abnormally. Tau in FTLD-tau, TDP-43 in FTLD-TDP, and FET proteins in FTLD-FET. It is in the second of these, FTLD-TDP, where new evidence is fueling a renewed interest in how the subtypes of FTLD-TDP are defined.
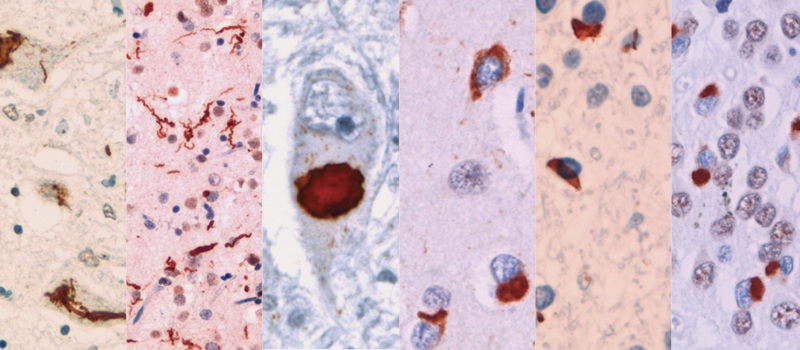
Since FTLD-TDP is marked by the accumulation of TDP-43, neuropathologists—natural splitters—have seen that the accumulation does not always look the same between patients. Under the microscope, the protein deposits vary in shape, in their location within cells, and in which cortical layers they favor. These differences have been formalized into subtypes from Type A to E, though A, B, and C account for the majority of cases. Some clinicians tend toward lumping. They see a shared toxic protein causing overlapping symptoms, and reason, logically, that drugs targeting TDP-43 accumulation ought to work regardless of how pathologists divide the patient population.
For years, most molecular studies either grouped all FTLD-TDP patients together or lacked the cohort size to say much about individual subtypes. Whether the pathological groups reflected biological differences was an open question, but answering it required large, well-characterized cohorts where subtypes could be examined separately. The group of Prof. Rosa Rademakers had them. The team approached the subtype question through three independent omics layers—genetics, epigenetics, and transcriptomics—each analyzed separately, each drawing on a different level of biology.

The genetic anchor
The genetics paper, published in Nature Communications and led by Dr. Cyril Pottier, reports the first genome-wide association study (GWAS) with enough power to examine FTLD-TDP subtypes individually. The two most frequently identified mutations causing FTLD-TDP, GRN and C9orf72, were already associated with specific subtypes; the GWAS extended that logic to identify common genetic variants more frequently seen in patients rather than in healthy controls. Unlike transcriptomic or epigenetic differences seen in patients, which could be either upstream or downstream of the disease process, genetic variants have no such ambiguity. They are present from birth, long before any pathology begins, and as such, if subtypes have different genetic risk profiles, the differences between them are fundamental. And that is what the study found—each subtype has a largely distinct genetic architecture.
FTLD-TDP Type A is dominated by genes involved in lysosomal function. The risk variants cluster around GRN, TMEM106B, and cathepsins like CTSB—all lysosomal genes. This converges with what is already known from rare mutations. Loss-of-function mutations in GRN invariably produce Type A pathology at autopsy, and GRN encodes progranulin, a protein critical for lysosomal health. The common variants identified in the GWAS—carried by many people in the population, each contributing a small increment of risk—tell the same story through a different mechanism. They don't eliminate progranulin but reduce its levels, nudging the lysosome toward dysfunction.
Type B showed enrichment for retrograde transport, with genes like VPS53 and DENND2A implicated. Type C diverged entirely. The first genome-wide significant risk locus identified for Type C was C19orf52 (also known as TIMM29), which mediates protein import into the mitochondrial inner membrane. Rare variant analysis uncovered RBPJL, a regulator of Notch signaling, a pathway not previously connected to FTLD-TDP genetics.
Only two genetic risk factors were shared across all subtypes: UNC13A, a known TDP-43 target gene, and TNIP1, which is involved in inflammation. These likely represent downstream consequences of TDP-43 accumulation rather than subtype-specific drivers—the cellular response to protein aggregation rather than its cause.
Epigenetic divergence
The second paper — the methylation study, led by Dr. Cristina Vicente and published in Molecular Neurodegeneration, approached the question from a different angle. DNA methylation patterns can shift with disease, but they also reflect developmental history and environmental exposures accumulated over a lifetime.
The striking finding was that FTLD-TDP subtypes share less than 10% of their epigenetic signatures. The methylation landscapes are almost entirely distinct. Within this small overlap, Type A patients and GRN mutation carriers shared the most, consistent with the genetic data showing lysosomal dysfunction as the common thread.
One gene emerged as particularly important: CAMTA1. In Type A patients, a region within CAMTA1 was consistently hypomethylated, and this correlated with reduced gene expression. CAMTA1 is already known to be regulated by TDP-43, so its dysregulation in FTLD-TDP makes biological sense. But the methylation changes were independent of TDP-43 levels, suggesting a "double-hit" mechanism: TDP-43 loss impairs CAMTA1 function, while parallel methylation changes make it worse. The findings reinforce the lysosomal connection because CAMTA1 plays roles in neuronal survival and development that intersect with lysosomal biology.
The splicing surprise
The third paper—the transcriptomics paper, led by Dr. Júlia Faura, published in Acta Neuropathologica, examined RNA splicing in frontal cortex tissue. TDP-43's primary function is regulating splicing, so analyzing splicing patterns in FTLD-TDP offers a window into how the disease disrupts cellular function.
Initially, Type A patients and GRN mutation carriers showed the most splicing alterations. But these diseases also cause the most neuronal death, and the team was analyzing bulk tissue—grinding up brain samples that mix surviving and dead cells. When they corrected for cell-type composition, accounting for the fact that Type A and GRN mutation carrier brains have fewer neurons, the picture shifted dramatically. Most splicing changes in these groups disappeared, revealing them as products of cell loss rather than specific disease mechanisms.
After correction, C9orf72 repeat expansion carriers showed the heaviest burden of splicing alterations. And Type C patients, who had appeared relatively quiet in the uncorrected analysis, showed a distinctive pattern—one that overlapped substantially with C9orf72 carriers.
This overlap was hard to explain: C9orf72 expansion carriers almost always show Type A or B pathology at autopsy, never Type C. Yet at the splicing level, they converge. Both groups showed aberrant splicing in NOTCH1 and four of its interaction partners. Notch signaling is essential for stem cell maintenance during development and for synaptic plasticity in adult brains.
The tantalizing finding is that both groups may share a neurodevelopmental component. C9orf72 repeat expansions have been shown to affect neural stem cell maintenance in prenatal mice. Research is ongoing to detect early effects of the mutation in teenagers from C9orf72 families—controversial work, because patient groups worry about stigmatization, but potentially important for understanding disease origins. Type C, meanwhile, is always sporadic and never familial, raising questions about whether environmental or developmental factors during early life might play a role. The disease trajectory could begin decades before symptoms appear.



Two visions, one goal
Does this molecular vindication of the splitters mean the lumpers are wrong? Not quite. As Prof. Rademakers says, both sides are right—they’re just asking different questions about the same patients.
If your target is TDP-43 itself—clearing it or preventing its accumulation—then treating FTLD-TDP as one disease makes sense. This approach mirrors current Alzheimer's strategies targeting amyloid. Drug companies favor broader patient populations. They aren’t eager to run gene therapy trials for a handful of patients in a rare subtype.
But if you want to treat the cause rather than the consequence, you must split. The pathways driving Type A (lysosomal dysfunction) differ from whatever drives Type C (potentially neurodevelopmental or metabolic factors). Progranulin gene therapy is already in clinical trials for GRN mutation carriers, with encouraging early results. But would it work in sporadic Type A patients who don't carry a GRN mutation? Probably not. However, lysosomal-boosting treatments could potentially benefit all Type A patients, because the GWAS shows lysosomal genes are the common thread.
Finding patients while they're still alive
None of this precision matters if you can't subtype patients before autopsy and right now, you can't. The Rademakers team is pursuing two ways to change that.
First, they're focusing on identifying biomarkers to distinguish FTLD-TDP patients from other FTLD forms. One approach involves developing antibodies to detect "cryptic peptides"—protein fragments that appear when TDP-43 mislocalizes from the nucleus and disrupts splicing. Clinicians could use these peptides and other biomarkers, detectable in spinal fluid and blood, to identify TDP-43 pathology in living patients.
However, while these biomarkers can reveal the presence of FTLD-TDP, they do not uncover its underlying pathological subtype. To go a step further, the Rademakers lab is building polygenic risk scores that capture complex gene interactions, aiming to create DNA-based tools that predict the most likely TDP-43 subtype in patients without known causal mutations.
If progranulin-targeting therapies succeed, accurate subtyping becomes critical. Patients with GRN mutations would benefit directly, but so might sporadic Type A patients whose lysosomal dysfunction mirrors GRN biology. Without a way to identify these patients during life, they'd never be enrolled in the right trials.
The path forward
All three papers used bulk tissue data which remains a limitation. The observed differences could partly reflect which cell types survived versus which died, not just what's going wrong inside surviving cells. The team is now conducting single-nuclei RNA sequencing to examine microglia, neurons, and other cell types separately, aiming to determine whether subtypes reflect different molecular failures inside surviving neurons or just different patterns of which neurons die first.
And there may be more subtypes than the pathologists recognized. A recently published collaboration performed unbiased clustering of protein profiles from the same patients—without imposing the A/B/C labels—and found the groups didn't align perfectly with neuropathological classifications. There might be four or more functionally distinct subtypes. The antibody stains capture something real, but perhaps not everything.
Darwin valued both hair-splitters and lumpers. In FTLD-TDP, the splitters are currently leading the charge toward precision medicine—their molecular work validating decades of careful neuropathological observation and pointing toward subtype-specific treatments. But the lumpers keep the door open for broad-spectrum approaches that could help patients regardless of pathological subtype. The path forward isn't about choosing sides. Understanding a disease this complex requires both the wide-angle lens and the high-resolution view.
Vinoy Vijayan